Redox biocatalysis offers attractive tools for the asymmetric synthesis of enantiopure molecules by combining high chemo- and stereo-selectivity usually with environmentally friendly process conditions. Exemplary reactions are (asymmetric) reduction of unsaturated compounds containing for instance C=C, C=O or C=N bonds (i). Several enzymes have been investigated within our research group and the development of reductive biocatalytic platforms has granted access to a broad variety of enantiopure α- and/or β-substituted carbonyl- and nitro-compounds, secondary alcohols, or primary and secondary amines. Our favorite enzymes include nicotinamide (NAD(P)H)-dependent ene-reductases, alcohol dehydrogenases and imines reductases. These enzymes are nicely complemented by transaminases and amino acid dehydrogenases, when novel functionalities are to be incorporated (amino group).
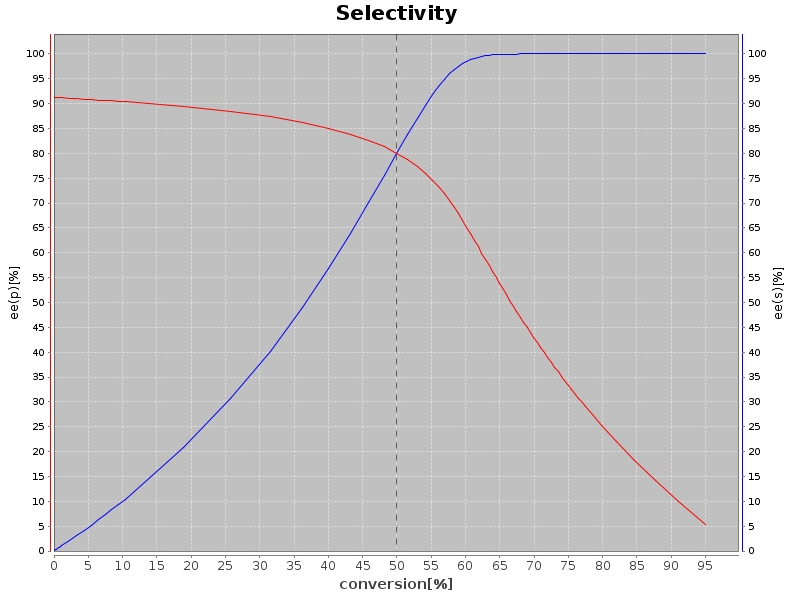
Oxidative reactions are also of interest, in particular if the biocatalyst shows high enantioselectivity on chiral molecules (ii), which results in kinetic resolution (KR) processes with racemates. Combining this step with (in situ) racemization increases the otherwise limited yield of the reaction to up to 100%, furnishing elegant dynamic kinetic resolution (DKR) strategies. Variations thereof have also been developed in our group (PIKAT). Oxidation reactions have been lately developed for alcohols, aldehydes or thio-compounds, employing among other alcohol oxidases and alcohol dehydrogenases, or Baeyer-Villiger monooxygenases.
A rising topic in our group is the valorization of renewables, aiming at combining naturally abundant carbon source with natural catalysts (iii). Here, P450 enzymes revealed highly efficient catalysts on fatty acids, for instance in decarboxylation reactions to deliver terminal alkenes (including dienes) or in C-H activation reactions (regioselective hydroxylation). Incorporation in multi-step enzymatic sequences then affords α-hydroxy, α-keto and (non-natural) α-amino-acids.
These redox enzymes typically rely on cofactors and/or coenzymes to transfer (reduction) or abstract (oxidation) electrons to/from a reactive substrate. A large number of them employ an external nicotinamide cofactor (NAD(P)H/NAD(P)+), often in conjunction with a flavin-coenzyme or a heme group, to shuttle electrons. For synthetic applications on large-scale, recycling strategies allow the use of (expensive) cofactor in catalytic amounts, while a (cheap) sacrificial auxiliary substrate serves as external electron source (reduction) or sink (oxidation). Although such technologies are well-established, they often suffer from an unfavorable environmental footprint impacted by the excess of co-substrate/product. We are working hard on developing alternative elegant strategies that allow recycling of cofactor, co-substrate or even reagent (oxidant). In some cases, we aim to affect equilibrium-reactions toward formation of the targeted products, while beneficial impact on overall atom-economy can also be observed (iv).
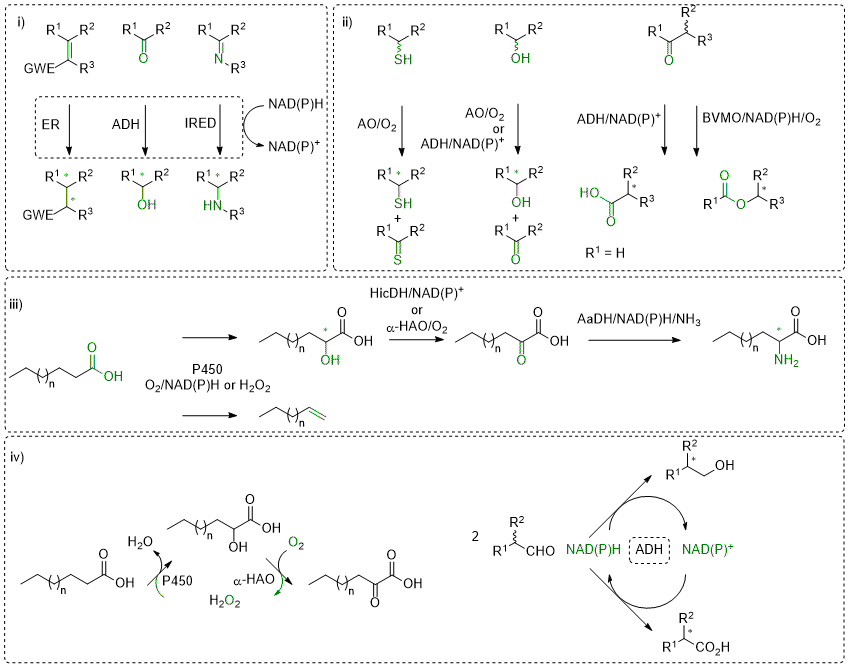
Scheme: i) Stereoselective reductive biocatalysis; ii) Oxidation reactions; iii) Valorization of fatty acids; iv) Recycling strategies. ER: ene-reductase; ADH: alcohol dehydrogenase; IRED: imine reductase; AO: alcohol oxidase; BVMO: Baeyer-Villiger monooxygenase; P450: P450 monooxygenase; HicDH: hydroxy acid dehydrogenase; α-HAO: hydroxy acid oxidase; AaDH: amino acid dehydrogenase.
Further reading:
i) Stereoselective reductive reactions (C=C / C=O / C=N reductions)
- Curr. Opin. Chem. Biol. 2018, 43, 97–105.
- Angew. Chem. Int. Ed. 2018, 57, 2864–2868.
- Adv. Synth. Catal. 2015, 357, 1655–1658.
- Angew. Chem. Int. Ed. 2008, 47, 9337–9340.
- J. Org. Chem. 2008, 73, 6003–6005.
- Angew. Chem. Int. Ed. 2007, 46, 3934–3937.
ii) Oxidation reactions (KR / DKR / PIKAT / BVMO)
- Adv. Synth. Catal. 2018, 360, 2742–2751.
- Adv. Synth. Catal. 2011, 353, 2354–2358.
- ACS Chem. Biol. 2016, 11, 1039–1048.
- J. Am. Chem. Soc. 2008, 130, 13969–13972
- Angew. Chem. Int. Ed. 2008, 47, 714–745
iii) Valorization of fatty acids
- Angew. Chem. Int. Ed. 2018, 57, 427-430.
- Org. Biomol. Chem. 2018, 16, 8030-8033.
- Catal. Lett. 2018, 148, 787–812.
- Eur. J. Org. Chem. 2016, 3473–3477.
- Angew. Chem. Int. Ed. 2015, 54, 8819–8822.
iv) Recycling strategies
- Angew. Chem. Int. Ed. 2018, 57, 427-430.
- Adv. Synth. Catal. 2018, 360, 2742–2751.
- Angew. Chem. Int. Ed. 2018, 57, 427–430.
- Green Chem. 2015, 17, 2952–2958.
- Angew. Chem. Int. Ed. 2014, 53, 14153–14157.

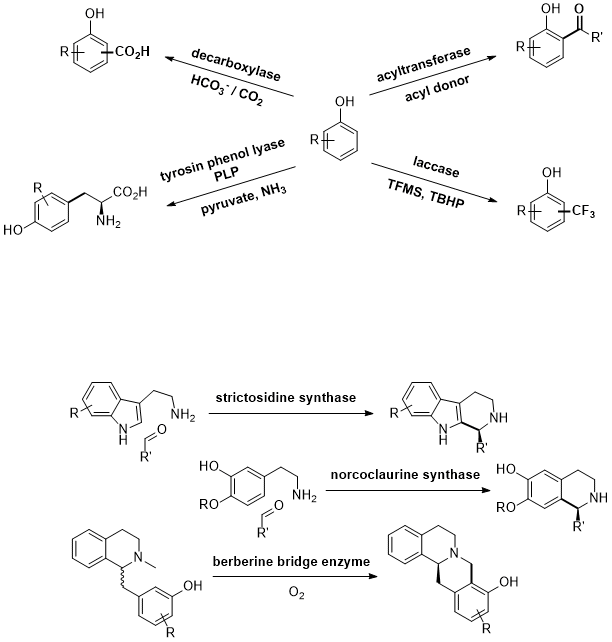
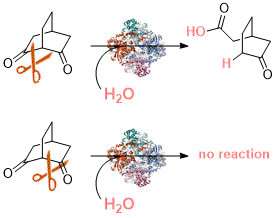

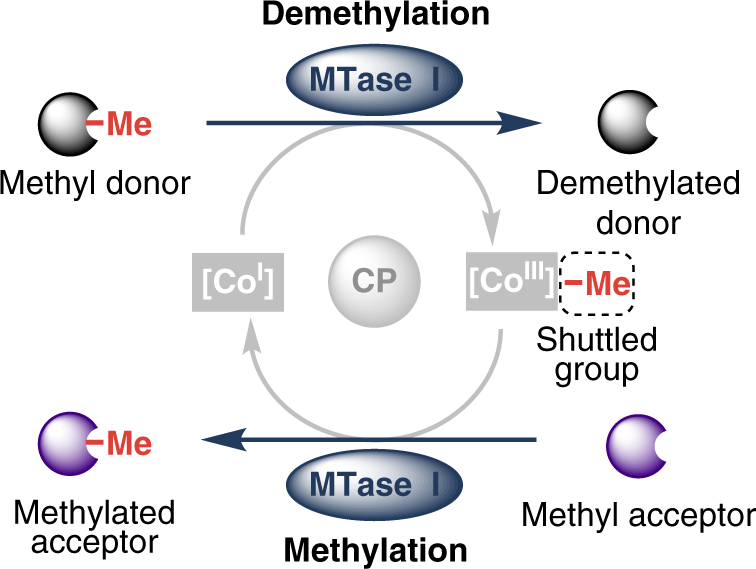
 .
.